





分子动力学模拟(详细教程 大聚合物插入生物膜中)
通过分子动力学模拟来探索不同理化性质 MPs(聚酯 PET、聚乙烯 PE、聚 丙烯酸酯 PP)与细胞膜的直接相互作用。
经文献调研,PET 微塑料包含多条聚对苯二甲酸乙二醇酯链,化学式为(C10H8O4)n/COC6H4COOCH2CH2O,每条链含约 100 个对苯二甲酸单体单元和 约 100 个乙二醇单体单元;PE 微塑料包含多条聚乙烯链,化学式为 (C2H4)n, 每条链含约 100 个乙烯单体单元;PP微塑料包含多条聚丙烯链,化学式为 (C3H6)n,每条链含约 100 个丙烯单体单元。
也就是
①PET100(C996H792O400)(邻苯二甲酸)
②PE100(C202H404) ethylene(乙烯)
③PP100(C301H602) propylene(丙烯)
通过 CHARMM-GUI 网站上的膜构建器获得 DPPC 双分子层。所得的脂 质双层由 256 个 DPPC 分子组成,膜的上下层对称分布。DPPC 双层的上小叶 和下小叶分别具有 128 个 DPPC 分子。DPPC 分子的亲水性头部向外,疏水性 尾部向内。DPPC 双分子层的初始结构在 GROMACS 53A6 力场下通过 100 ns 的分子动力学模拟进行结构优化。
一.数据生成charmm-gui/Materials Studio
https://charmm-gui.org/?doc=input/polymer
这里分为两种获取方式,因为gui网站生成蛋白质类物质特别的方便,因此DPCC的生成我们选择gui网站下载。聚合物方面我们选择选用MS进行生成并优化,因为gui网站下载的聚合物没有进行优化,在后续拓扑中缺少二面角等信息,因此我们聚合物选择MS进行生成。
1. DPCC生成
参考:https://www.bilibili.com/read/cv15787466/
到这个网站上下载数据
在这里我们要生成256个DPPC分子组成的的模拟双分子层,膜的上下层对称分布。
点击这里(这个注册需要一点时间,需要你填好信息以后,官方对你的信息进行审核,审核过程1天到半个月不等,官方审核完会把你的账号密码发送到你的邮箱里)
a 使用说明
依次点击Input Generator –> Membrane Builder –> Bilayer Builder,就会出现下图,网页下拉就能看到Protein/Membrane System以及Membrane Only System两个模块。
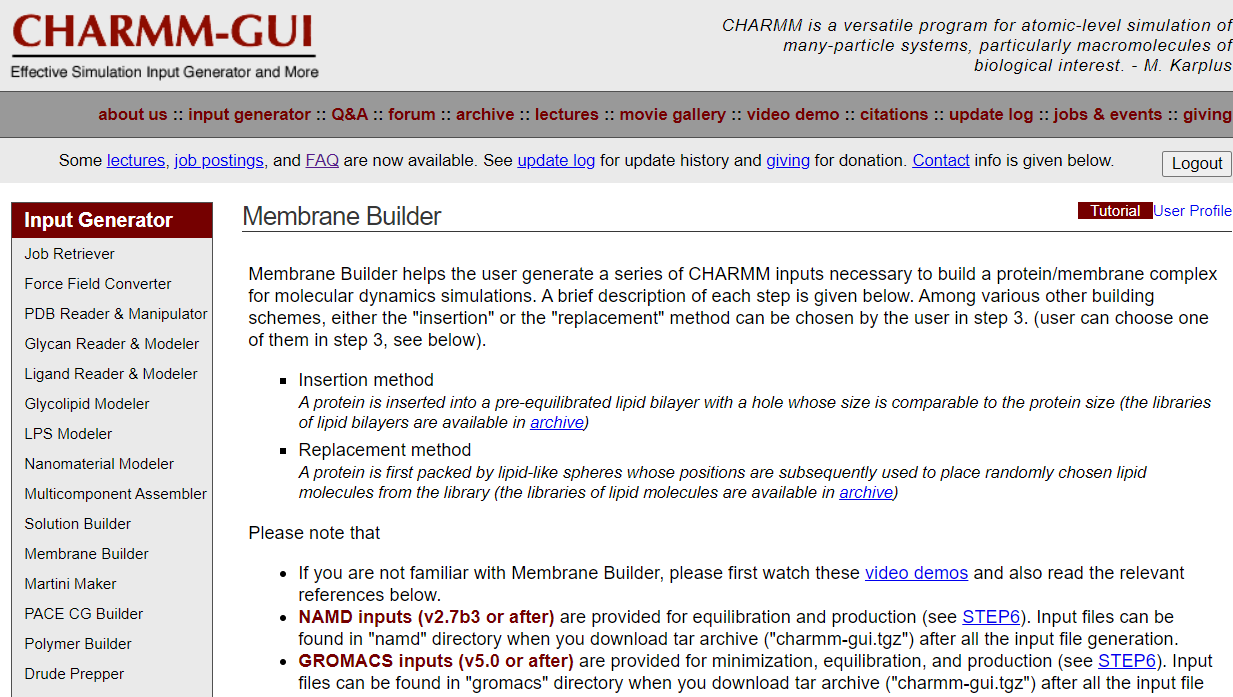
Membrane Builder可以帮助用户生成一系列charmm输入,以构建用于分子动力学模拟的蛋白质/膜复合物。
- 注意在Please note that下面有三句话:
1.a homogeneous lipid bilayer can be built with DMPC, DPPC, DOPC, POPC, DLPE, and POPE.(DMPC、DPPC、DOPC、POPC、DLPE和POPE可形成均质脂质双分子层)
2.a heterogeneous lipid bilayer can be built with 434 different lipid molecules (see lipid list).(非均相脂质双分子层可以由434种不同的脂质分子组成(看lipid list))
3.the heterogenous Membrane Builder can be used for a homogeneous lipid bilayer (only using the replacement method).(非均质膜构建模块可用于均质脂质双分子层构建)
b 构建DPPC膜
1)点击Membrane Only System
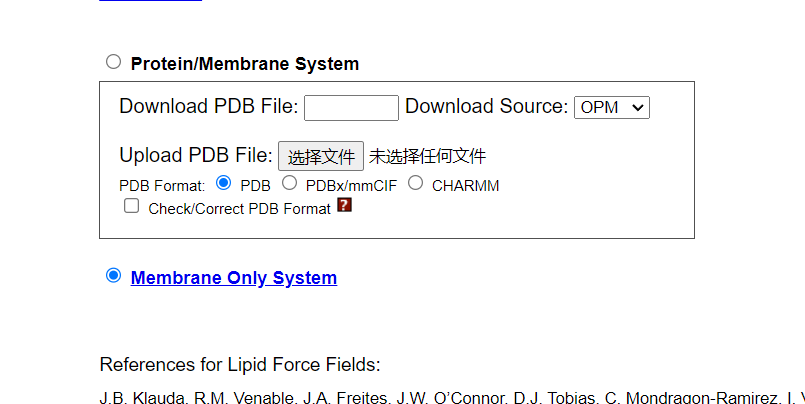
2)点击Next Step: Select Lipids,就会出现下图所示页面:
出现一行红字:”Homogeneous Lipid”选项不在支持,可以使用”Heterogeneous Lipid”来构建Homogeneous Lipid。所以我们直接
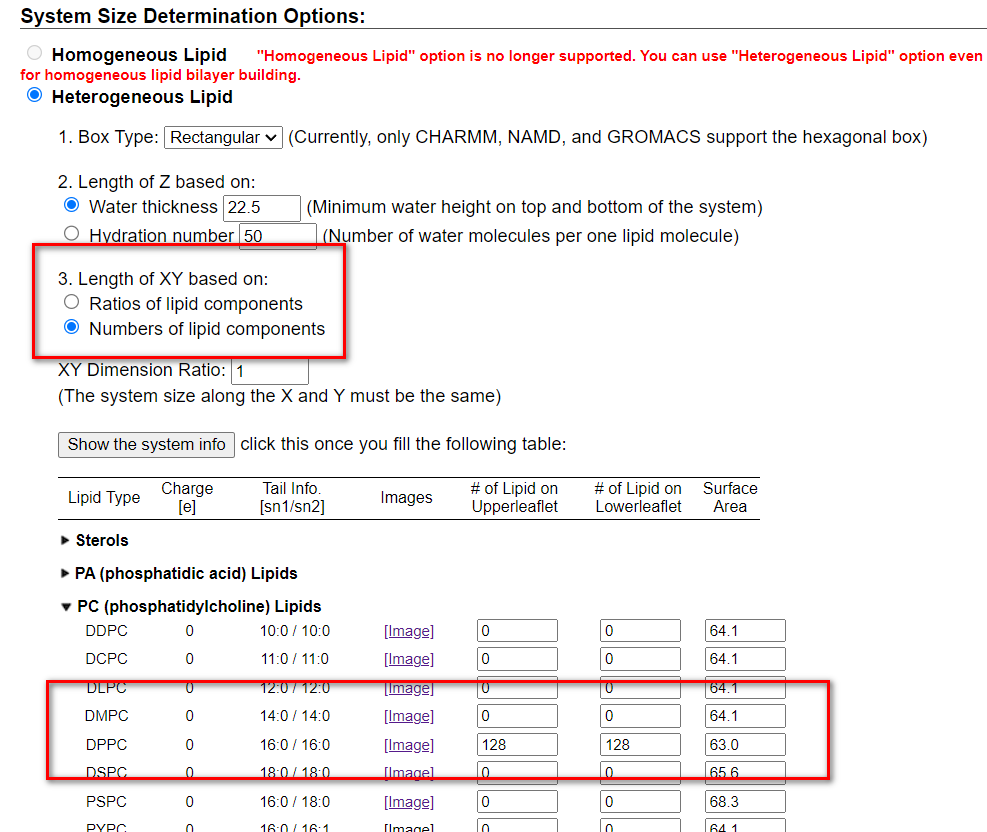
3)点击Heterogeneous Lipid,出现下图
一般不需要变动,因为我们需要构建256个DPPC分子,因此我们在3.中选择Number of lipid components ,上面指的是比例,如果选择比例,我们在下面选择构成比例时就要保证加起来等于100。我们这里选择DPPC分子,上下各128个分子。然后点击show the system info然后一直下一步就行
2. MS生成优化后的聚合物
使用 Materials Studio8.0 软件生 成 并 分 别 构 建 优化 PET100(C996H792O400)、PE100(C202H404)和 PP100(C301H602)的结构。
这里只展示PE的生成过程
a. 开始构建
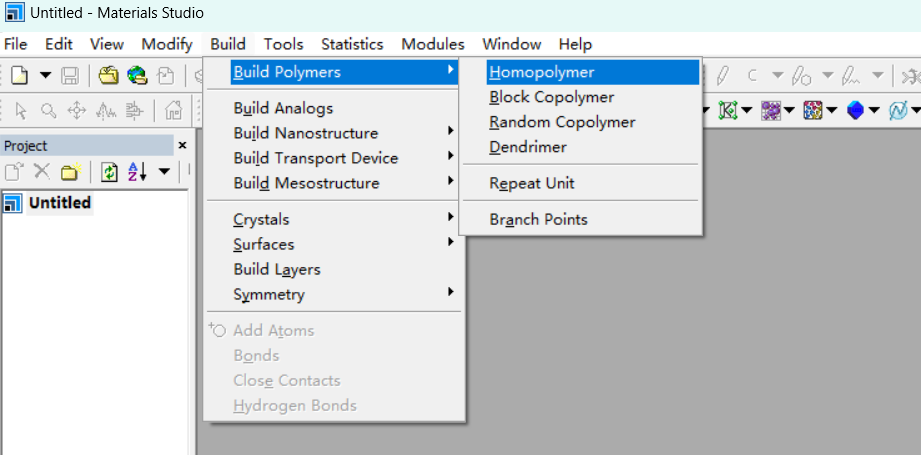
聚合度为100的PE链
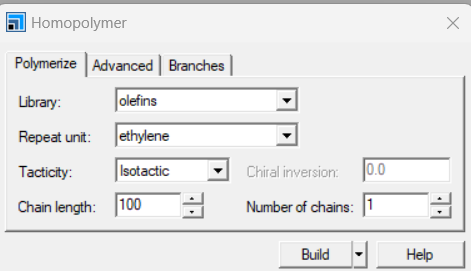
b对单链进行结构优化
选择Forcite模块中的calculation。Setup栏选择Geometry Optimization, 选择Algorithm类型,针对聚合物单链,本人一般简单的smart,Energy选择力场,针对PE我选用pcff力场,Job control选择并行数,一般调到最大。
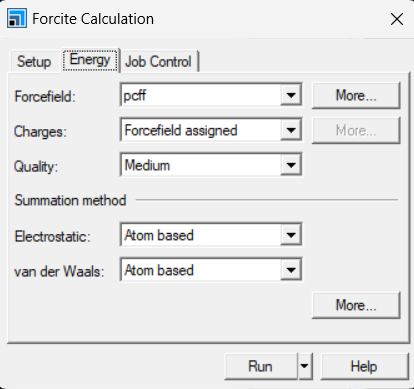
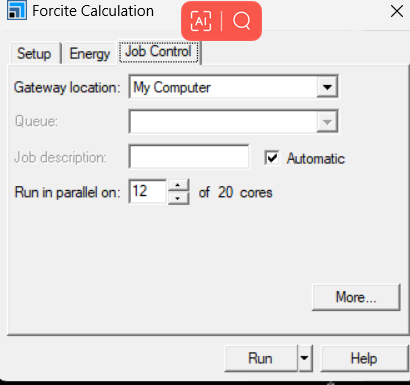
c 建盒子
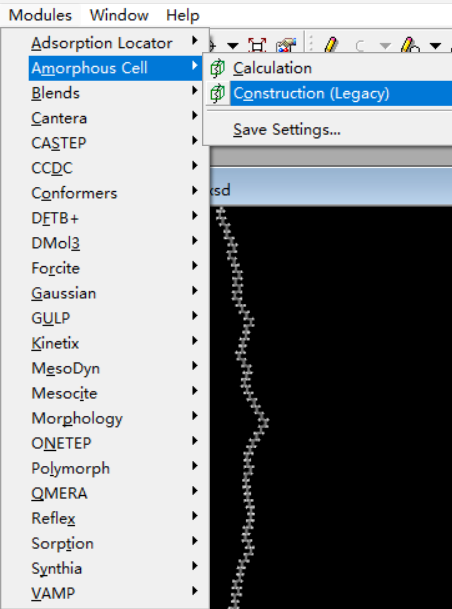
AC模块下的construction(Legacy),确保优化后的PE单链界面处于打开状态,进行add,选择cell type,设置密度,盒子大小等。这里我选用的是confined layer,至于和periodic有什么区别,自己可以进行尝试。设置numbers of configuration框为1,要不然你电脑会特别卡,然后取消下面两个勾选框。Setup栏注意力场要和之前的力场保持一致,后续的所有过程都要检查力场。
d 进行能量最小化
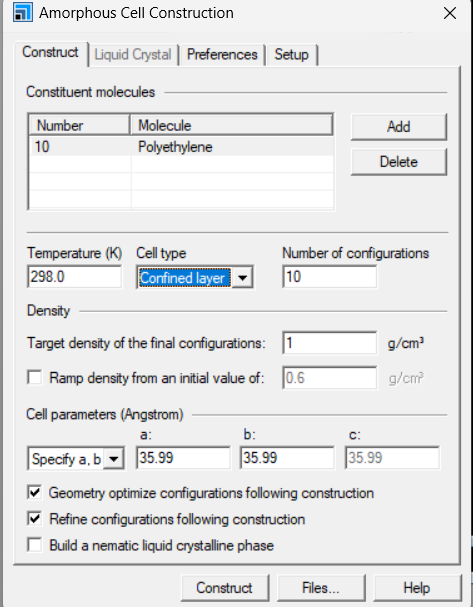
和步骤b类似,为了后续的弛豫过程不报错,在这里可以多进行几次结构优化,Steepest descent, Conjugate gradient, Quasi-Newton各来一次。
e 弛豫。
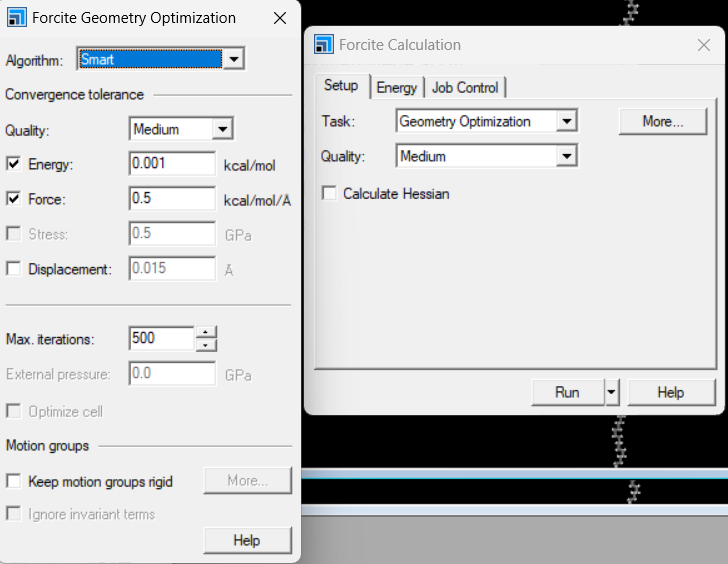
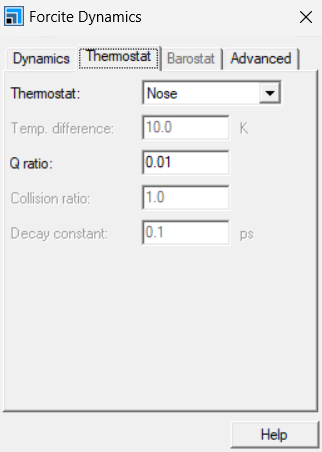
同理,将需要优化后的结构显示与主屏幕,FC中选择Dynamcis,检查力场,在more中选择系综,个人在MS中一般用NVT,弛豫时间100ps。热浴为经典的Nose-Hoover,确认力场,其余默认,弛豫时间一般较长,看电脑性能。
e 导出模型
点击打开最后的xtd文档,然后点击export导出你想要的格式就可以了,这里我们导出PDB格式
二 GROMACS的使用
参考文章
https://mp.weixin.qq.com/s/LiWO1F199Pxy3eLUFN7jhA
https://jerkwin.github.io/2017/09/14/GROMACS非标准残基教程1-修改力场与增加残基/
官方教程文档:http://www.mdtutorials.com/gmx/membrane_protein/03_solvate.html
参数内容:https://www.compchems.com/gromacs-mdp-file-parameters/#mdp-file-for-npt-equilibrations
参数官方文档:https://manual.gromacs.org/current/user-guide/mdp-options.html
中文官方教程(感觉是偏原理解释,没有代码操作教程):https://jerkwin.github.io/GMX/GMXman-1/
1 拓扑文件准备:
只有构型文件是不够的,因为构型文件里面只有这些分子的各个原子的顺序和坐标,并没有它们之间的连键情况等,故进行模拟还需要体系中各成分的拓扑文件。对于蛋白质的拓扑文件,可以在gromacs中通过命令pdb2gmx来生成,因为gromacs中包含很多种氨基酸的拓扑文件和转换脚本。对于其他分子的拓扑文件,在下载构型文件时,往往会包含相应的itp文件,即为分子的拓扑文件。水分子和离子等小分子的拓扑文件在gromacs的立场里面都有,可直接调用。
2 参数文件
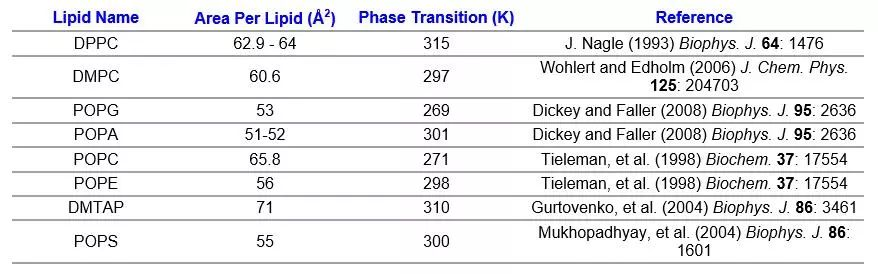
一般在gromacs tutorial中都可以直接下载(后缀为mdp的文件,包括ions.mdp, minim.mdp, nvt.mdp, npt.mdp, md.mdp),然后设置一些参数,包含模拟方法、时间、步长、输出时间间隔、键参数设置、相邻单元、静电、温度、压力、速度、周期边界条件等,有时还包括一些限制、电场、力等。对于温度的设置,应当设置在磷脂的相转变温度以上,如下是一部分磷脂的相转变温度。
也可以
3 流程
a.制作拓扑文件(化合物较小)
注意如果你是重复型的化合物坚持用tpp也可以得到拓扑但是会在后面出现问题详情在3.7有讲解
tppmktop是获得有机分子OPLSAA力场拓扑文件的好工具, 但也存在一定的不足. 一是不支持周期性分子, 二是提供的网络服务同时只能运行一个任务. 为此, 我觉得还是有必要自己做一个简单OPLSAA力场拓扑生成器GMXTOP, 一则可用于理解力场拓扑的生成方法, 二则可用于检查其他方法得到的拓扑是否合适.
参考文章:https://jerkwin.github.io/2017/09/14/GROMACS非标准残基教程1-修改力场与增加残基/
将你上面得到的聚合物pdb文件提交到这个网站上http://erg.biophys.msu.ru/erg/tpp/
然后等待一到三分钟就能得到该文件的itp和rtp文件,这两个文件是力场的拓扑文件
这个是PDB文件,这里有一个残基的名称
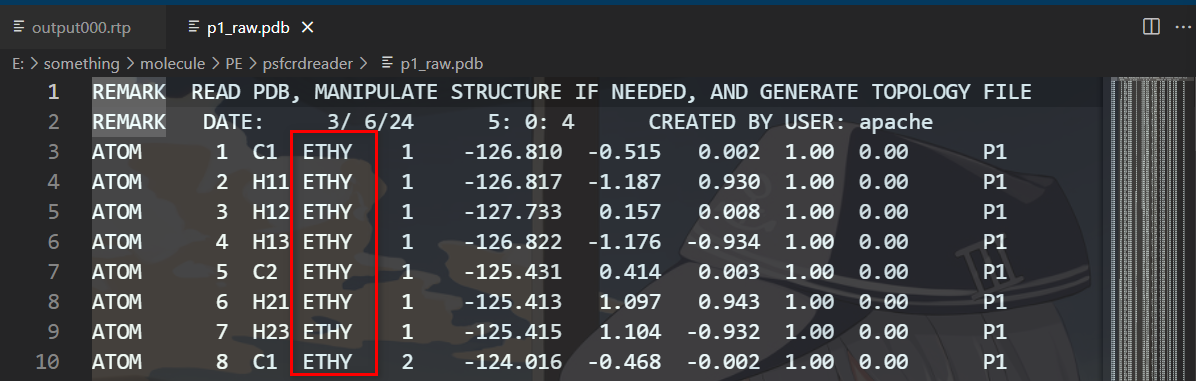
然后把导出的rtp和itp文件中的这个位置改成你残基的名称
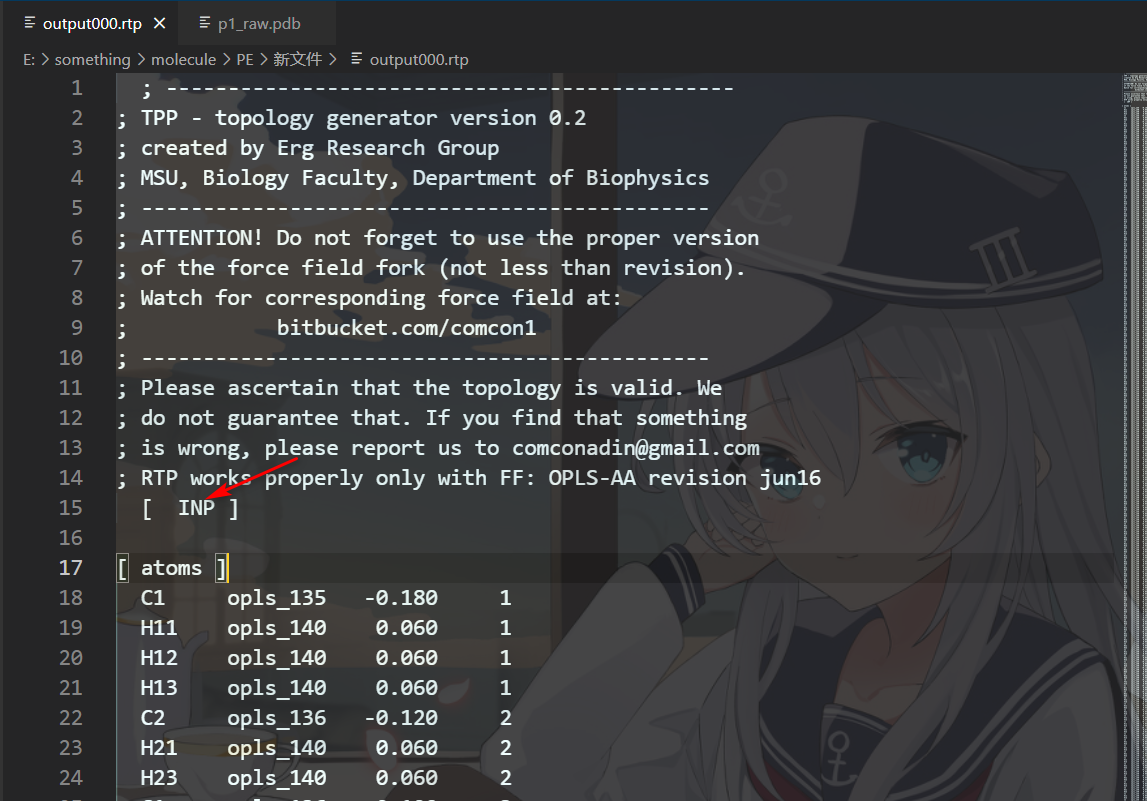
rtp和ipt文件编辑完成以后,把文件复制到/gmx2024.1/share/gromacs/top/oplsaa.ff 的文件夹下,然后进行下面的操作。
b.制作拓扑文件(化合物较大并重复)
参考文章:http://bbs.keinsci.com/thread-41715-1-1.html
进入网站:https://cloud.hzwtech.com/web/personal-space/auto-ff/all-atom
上传你的文件并选择你想要的立场等参数就可以生成对应的文件辣,这个网站实在是太方便了
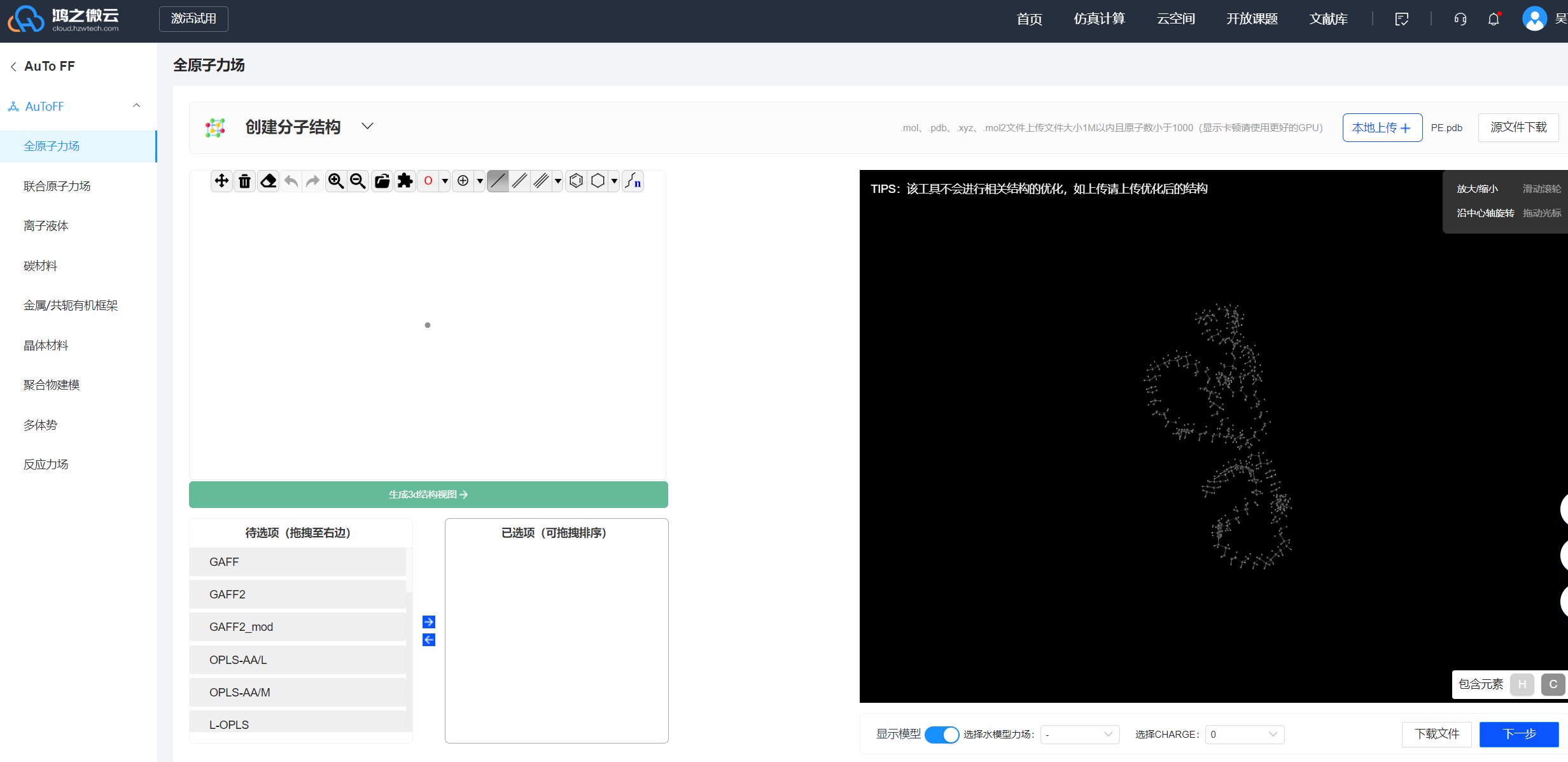
c. 构造体系
(1)将多肽的pdb文件转换成gro文件,同时生成其拓扑文件。(如果你进行了上面拓扑这一步可跳过,直接使用上面生成的gro文件)
1 | gmx pdb2gmx –f protein.pdb –o protein.gro |
在模拟蛋白质的时候,要考虑氢原子、蛋白质的两端以及带电残基等,可以用-ingh(忽略氢原子), -ter(选择多肽的两端氨基和羧基的带电情况), -inter(交替分配电荷状态)来实现。在这一步中将会选择立场,蛋白质的两端。
完成该步之后生成protein.gro, topol.top, posre.itp,其中topol.top为多肽的拓扑文件,在其中引入了立场,多肽的原子性质,连键情况、形成的角等,并且引入了水和离子的拓扑文件;posre.itp是蛋白质的重原子,用于后面对蛋白质进行位置限制。实际上总的拓扑文件里只要包含立场、各个分子的结构性质即可,无论是直接写在里面也好,还是通过include来引入也好。
(2)给生成的多肽构型定义一个盒子(根据整个体系的盒子尺寸来构造),并且将多肽放在合适的位置上。
1 | gmx editconf –f protein.gro –o pro_newbox.gro –box 长 宽 高 -center 长 宽 高 |
也可以用我的默认参数
1 | gmx editconf -f protein.gro -o pro_newbox.gro -c -d 1.0 -bt cubic |
盒子的大小要取决与膜的大小(见下面步骤)。为了将多肽放在膜上方来进行模拟,所以在此定义的位置应该与后面生物膜的位置有一定距离。该步生成pro_newbox.gro。
(3)给生物膜定义一个同样的盒子,并放置在相同的位置上。
1 | gmx editconf –f bilayer.pdb –o bilayer_newbox.gro –box 长 宽 高 -center 长 宽 高 |
注意双层膜的位置不要与多肽重叠。并且盒子的长和宽要与膜的大小相匹配,否则后面加水后磷脂的疏水尾部也将暴露在水中。如果不知道下载的膜的长宽,可以先根据经验定义一个盒子,然后用vmd打开后,输入指令pbc box,就可以显示盒子的框架了,看看是否适合,不合适就进行调整。关于vmd的使用(此处VMD教程在后面),在本文的最后会有详细介绍。如果下载的磷脂膜模型较小,可以用cat命令将两个放在一起,具体使用方法类比下面cat命令介绍。或者用如下命令:
1 | gmx genconf -f bilayer_newbox.gro -nbox 2 1 1 -o bilayer_big.gro |
其中-nbox后面的数字分别是在x、y、z方向上复制的倍数。得到的新的磷脂膜最好经过一段时间的平衡,使其连接处更加连贯。
(4)将多肽与膜放在一起,并且添加水分子。
1 | cat pro_newbox.gro bilayer_newbox.gro > whole.gro |
其中这里只是把两个分子强行融合到一个盒子里面,难免会有误差比如这样

因此我们需要通过软件来调整PE的位置,因为gro文件格式如下,我们在Excel里面就可以调整我们想要调整分子的坐标,但是这样不直观,每次的改变都不能看到,因此我们是用VMD进行调整。具体调整过程请看4.a
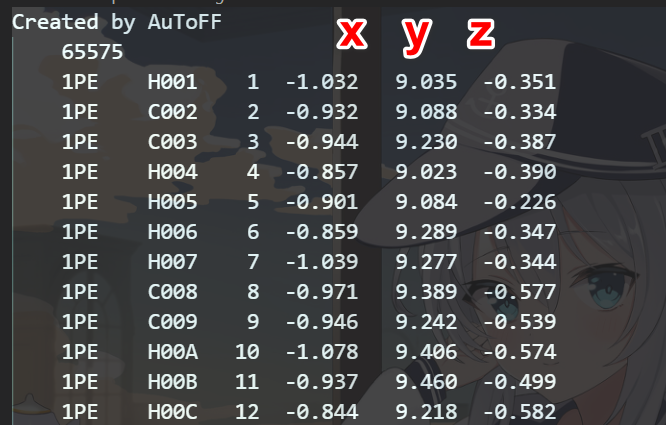
我们把CHARMM-GUI生成的DPPC文件中的top文件包含进我们上面的top文件里,
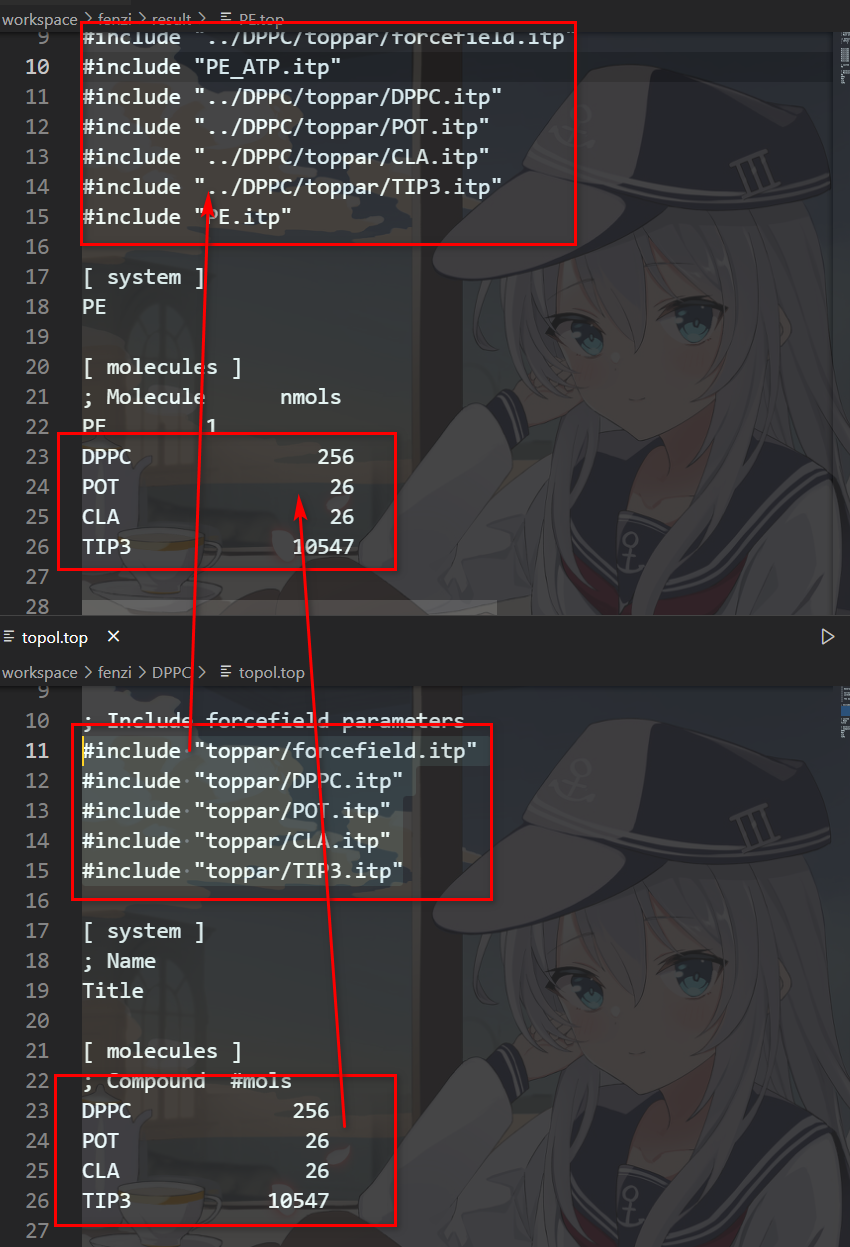
然后一定要按顺序导入文件
1 | #include "../DPPC/toppar/forcefield.itp" |
第一第二行是分子的定义,一定一定要把分子定义放在前,不然会报Invalid order for directive 的错误。
什么是分子的定义,含有[atomtypes]标签的就算作分子的定义
这段实在没懂咋设置我们实验的值在加水之前,应当将vdwradii.dat中的C的值从0.15改为0.375,这样是为了防止加水后生物膜的空隙中也有水,如果此时还有极少量的水分子在膜空隙中,可以用写字板等直接编辑生成的gro文件,删除其中的水分子,具体步骤祥见下面的vmd部分。删除水分子之后,别忘了top文件里面相应的水分子数目要减少。
注意:如果之前DPPC加过水了,这里就不用加水
1 | gmx solvate –cp whole.gro –cs spc216.gro –o sol.gro –p topol.top |
d 开始模拟
(1)添加约束力(可跳过)(后面也有约束力)
1 | gmx genrestr -f sol.gro -o strong_posre.itp -fc 100000 100000 100000 |
选择整个系统0
在用于最小化的 mdp 文件,添加一行“ Definition =-DSTRONG _ POSRES”来使用这些新的位置约束。
inflategro.pl下载路径如下:http://www.mdtutorials.com/gmx/membrane_protein/Files/inflategro.pl
原文语句:perl inflategro.pl system.gro 4 DPPC 14 system_inflated.gro 5 area.dat
- 4-应用的缩放因子。值 > 1表示通货膨胀,值 < 1表示收缩/压缩
- DPPC-用于脱皮的油脂的残留名称
- 用于搜索要删除的脂质的截止半径(单位: Å)
- 每个油脂计算面积的5个网格间距(也以 Å 为单位)
- Dat-输出文件,包含每个脂质的面积信息,可用于评估结构的适应性
因此语句为
1 | perl inflategro.pl sol.gro 1 POPE 12 system_inflated.gro 5 area.dat |
然后会被删除一些原子因此我们需要再topol.top中的molecules 模块更改修改后的原子数目。
(2)能量最小化文件准备
在每个不同的体系下需要不同的参数,这里面以蛋白质为例,你可以根据你想要的参数进行调整
1 | ; LINES STARTING WITH ';' ARE COMMENTS |
1 | gmx grompp –f minim.mdp –c system_inflated.gro –p topol.top -r system_inflated.gro –o min.tpr |
如果这里又报错可以强行跳过
-maxwarn 6
输出文件包括构型文件(min.gro)、日志文件(min.log)、轨迹文件(min.trr、min.xtc)。可以通过修改参数文件内的输出时间间隔来控制输出,最好不要过于细致(占内存较大),也不要过于粗略(不利于分析)。
如果这一步报错:No default Proper Dih. types 移步到3.3
然后运行
1 | gmx mdrun -v -deffnm min |
如果系统没有收敛则移步:http://www.mdtutorials.com/gmx/membrane_protein/05_EM.html
如果报错:The [molecules] section of your topology specifies more than one block of a [moleculetype] with a [settles] block. 移步3.6
输出文件包括构型文件(min.gro)、日志文件(min.log)、轨迹文件(min.trr、min.xtc)。可以通过修改参数文件内的输出时间间隔来控制输出,最好不要过于细致(占内存较大),也不要过于粗略(不利于分析)。
(3)建立index文件/约束力
1 | gmx make_ndx -f min.gro -o index.ndx |
在建立过程中,会选择分组,比如,我们可以将水和离子分为一组,根据提示可以将水和离子前面的序号用“|”连接即可,如14|16。
1 | PE_DPPC |
然后用q退出
热平衡和压力平衡,为了防止在NVT和NPT的时候蛋白复合物爆开,所以要加一个力·的限制,首先构建配体的力限制
1 | #创建索引文件 |
给增加1000的三个方向的力
1 | gmx genrestr -f min.gro -n index_force.ndx -o force.itp -fc 1000 1000 1000 |
之间插入位置
1 | ; Include ligand Position restraint file |
(4)nvt模拟
nvt.mdp
1 | define = -DPOSRES -DPOSRES_FC_LIPID=1000.0 -DDIHRES -DDIHRES_FC=1000.0 ; position restrain the protein and ligand |
注意tc-grps = PE_DPPC POT_CLA_TIP3的热浴组合温度需要改成自己的,这个体系用的常温300K,跑的时间为100ps
1 | gmx grompp -f nvt.mdp -c min.gro -r min.gro -p topol.top -n index.ndx -o nvt.tpr |
nvt平衡中通常使用的热浴为Berendsen或者V-rescale,使最终温度能基本达到稳定。
类似能量最小化,只须将脚本中的min改为nvt即可。输出文件比能量最小化多了一个断点文件(nvt.cpt)
完事以后我们继续对产生的文件进行
1 | gmx mdrun -v -deffnm nvt |
(5)npt模拟
nvt模拟之后温度基本达到稳定,但是还需将模拟盒子根据设定的压力进行平衡模拟以松弛磷脂膜,因此进行npt模拟只需限制蛋白质即可。但还应注意的是,在npt模拟中一般使用Nose-Hoover热浴,与nvt中的两个热浴不同,该热浴允许比较大的涨落,因此不适用于nvt模拟。对于压力运用Parrinello-Rahman方法,由于模拟盒子中有磷脂膜,所以我们在此使用semiisotropic,与膜平行的两个方向为一个压力,与膜垂直的方向为一个压力。
npt.mdp
1 | title = Protein-ligand complex NPT equilibration |
1 | gmx grompp –f npt.mdp –c nvt.gro -r nvt.gro –t nvt.cpt –p topol.top –n index.ndx –o npt.tpr |
将脚本中nvt改为npt,即可提交任务。
1 | gmx mdrun -v -deffnm npt |
(6)md模拟
平衡了温度和压力之后,开始较长时间的md模拟,模拟过程与npt类似。
如果计算结束以后,觉得仍未平衡,或者想要更长时间的模拟,可以进行续算,一种方法是类似上面,只是将md.gro作为初始构型,进行md模拟;另一种方法是延长计算时间,用-extend 10000延长10000ns。
md.mdp
title = Protein-ligand complex MD simulation
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000000 ; 2 * 50000000 = 100000 ps (100ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 1000 ; save coordinates every 200 ps
nstvout = 1000 ; save velocities every 200 ps
nstxtcout = 1000 ; xtc compressed trajectory output every 200 ps
nstenergy = 1000 ; save energies every 200 ps
nstlog = 1000 ; update log file every 200 ps
; Bond parameters
continuation = yes ; continuing from NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds to H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighbor searching and vdW
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 20 ; largely irrelevant with Verlet
rlist = 1.2
vdwtype = cutoff
vdw-modifier = force-switch
rvdw-switch = 1.0
rvdw = 1.2 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
rcoulomb = 1.2
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = PE_DPPC POT_CLA_TIP3 ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction is not used for